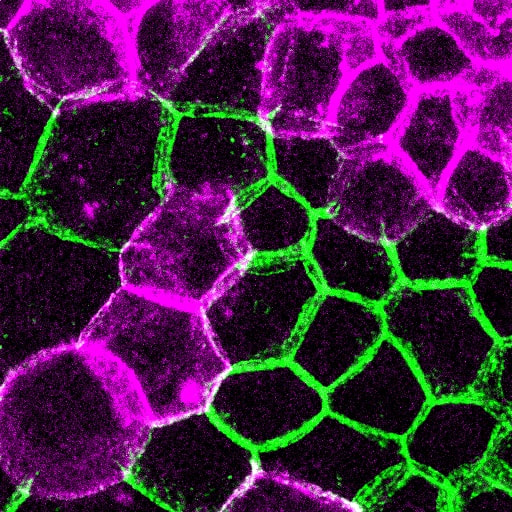
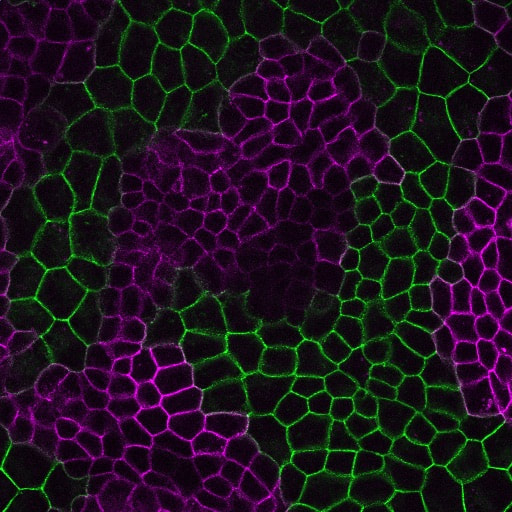
How is cell-cell adhesion regulated?
Cell-cell cohesion is a defining feature of multicellular organisms, and a number of disease states—from cancer to heart disease-- are driven by defects in the structural “Velcro” that holds cells to one another. It is widely viewed that the cadherin/catenin adhesion complex is a master regulator of this adhesive “Velcro.” This complex comprises a transmembrane cadherin component that mediates calcium-dependent homophilic recognition and a number of associated catenins (e.g., alpha, beta, gamma and p120-catenins) that link cadherins to the underlying cytoskeleton. Over the last 25 years, our field has largely established the requirement of each component to intercellular adhesion (and adhesion-dependent processes) in systems ranging from cell culture to loss-of-function studies in flies, worms and mice. What has emerged is a “chain-of-adaptors” model of cell-cell adhesion where cadherin receptors effectively join the cytoskeletons of adjacent cells through a cadherin/b-catenin/a-catenin/F-actin linkage. While live cell imaging studies have revealed the dynamic nature of cell-cell adhesions, we know comparatively little about the signals and modifications that control this dynamic behavior—and what constitutes “strong” versus “weak/fluid” adhesion at the molecular level. We are currently leveraging our proteomics analyses with a multi-disciplinary team of collaborative investigators to determine how the cadherin/catenin complex is regulated, which will have broad implications for intercellular adhesion across diverse cell types, and may be relevant to pathophysiological conditions that impact epithelial barrier function, tissue size and integrity
Cell-cell cohesion is a defining feature of multicellular organisms, and a number of disease states—from cancer to heart disease-- are driven by defects in the structural “Velcro” that holds cells to one another. It is widely viewed that the cadherin/catenin adhesion complex is a master regulator of this adhesive “Velcro.” This complex comprises a transmembrane cadherin component that mediates calcium-dependent homophilic recognition and a number of associated catenins (e.g., alpha, beta, gamma and p120-catenins) that link cadherins to the underlying cytoskeleton. Over the last 25 years, our field has largely established the requirement of each component to intercellular adhesion (and adhesion-dependent processes) in systems ranging from cell culture to loss-of-function studies in flies, worms and mice. What has emerged is a “chain-of-adaptors” model of cell-cell adhesion where cadherin receptors effectively join the cytoskeletons of adjacent cells through a cadherin/b-catenin/a-catenin/F-actin linkage. While live cell imaging studies have revealed the dynamic nature of cell-cell adhesions, we know comparatively little about the signals and modifications that control this dynamic behavior—and what constitutes “strong” versus “weak/fluid” adhesion at the molecular level. We are currently leveraging our proteomics analyses with a multi-disciplinary team of collaborative investigators to determine how the cadherin/catenin complex is regulated, which will have broad implications for intercellular adhesion across diverse cell types, and may be relevant to pathophysiological conditions that impact epithelial barrier function, tissue size and integrity
Adhesion-signaling dysregulation and disease: It’s not only about cancer!
Generally speaking, the cell-cell adhesion complex we study is so fundamental that loss-of-function mutations are associated with developmentally devastating consequences (i.e., embryonic lethality). Perhaps it is for this reason that most of us assumed that mutations in this complex could only be tolerated focally, as in cancers. But recent genome-wide associations studies appear to be telling us otherwise. Specifically, we’ve learned that a developmentally dispensable form of a-catenin, known as aT-catenin, is highly restricted to expression within cardiomyocytes of the heart/lung and a subset of cell-types in the brain, which may be relevant to its numerous associations with diseases ranging from asthma to autism. Using a fully viable aT-catenin knock-out mouse, we’ve been able to validate the contribution of this protein to the development of asthma, and are currently interested in uncovering the contribution of a non-canonical cell type (heart or brain?) to this disease.
Generally speaking, the cell-cell adhesion complex we study is so fundamental that loss-of-function mutations are associated with developmentally devastating consequences (i.e., embryonic lethality). Perhaps it is for this reason that most of us assumed that mutations in this complex could only be tolerated focally, as in cancers. But recent genome-wide associations studies appear to be telling us otherwise. Specifically, we’ve learned that a developmentally dispensable form of a-catenin, known as aT-catenin, is highly restricted to expression within cardiomyocytes of the heart/lung and a subset of cell-types in the brain, which may be relevant to its numerous associations with diseases ranging from asthma to autism. Using a fully viable aT-catenin knock-out mouse, we’ve been able to validate the contribution of this protein to the development of asthma, and are currently interested in uncovering the contribution of a non-canonical cell type (heart or brain?) to this disease.
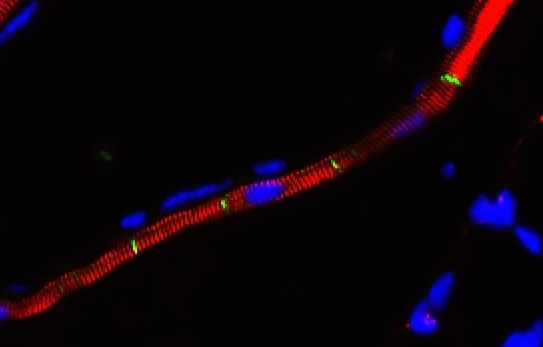
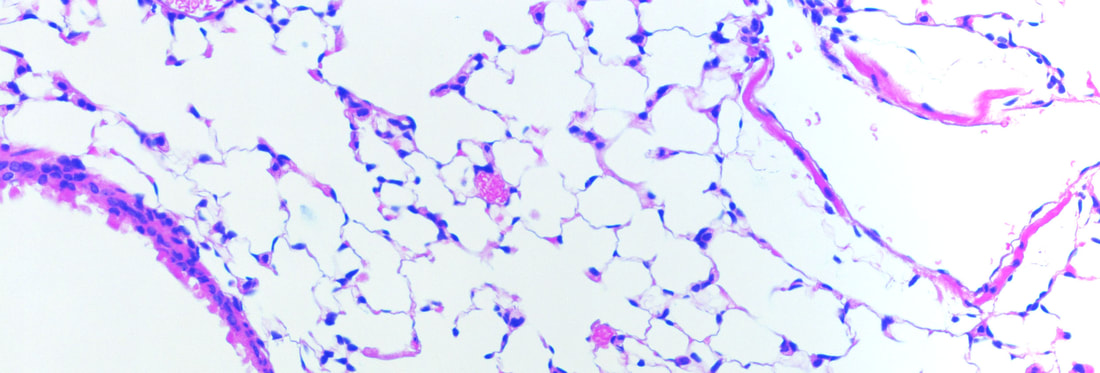
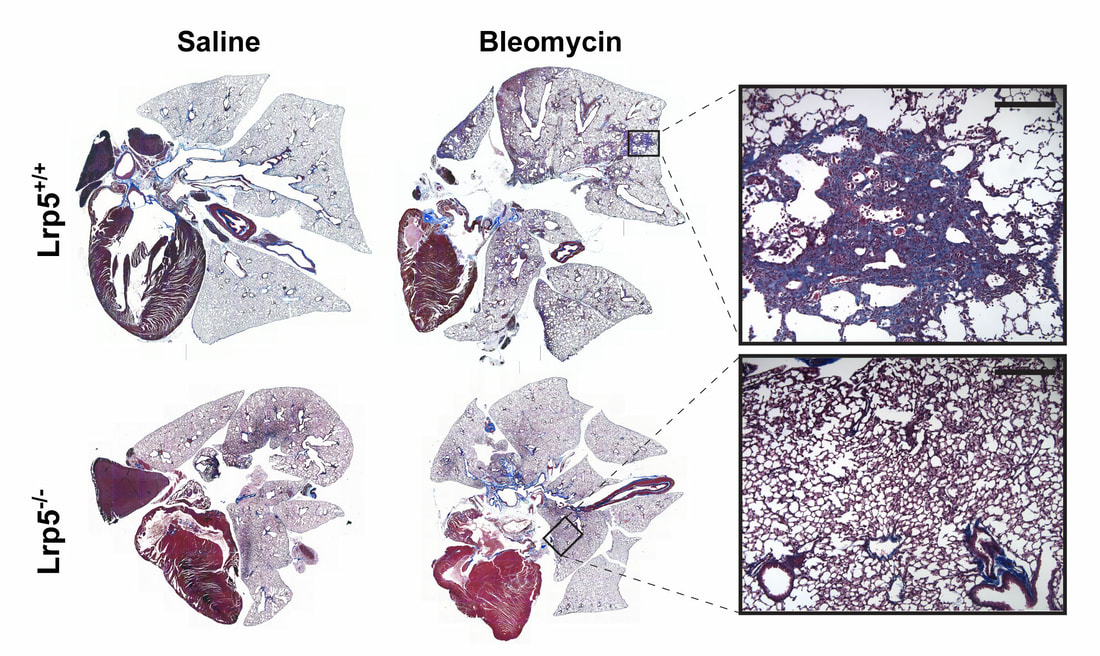
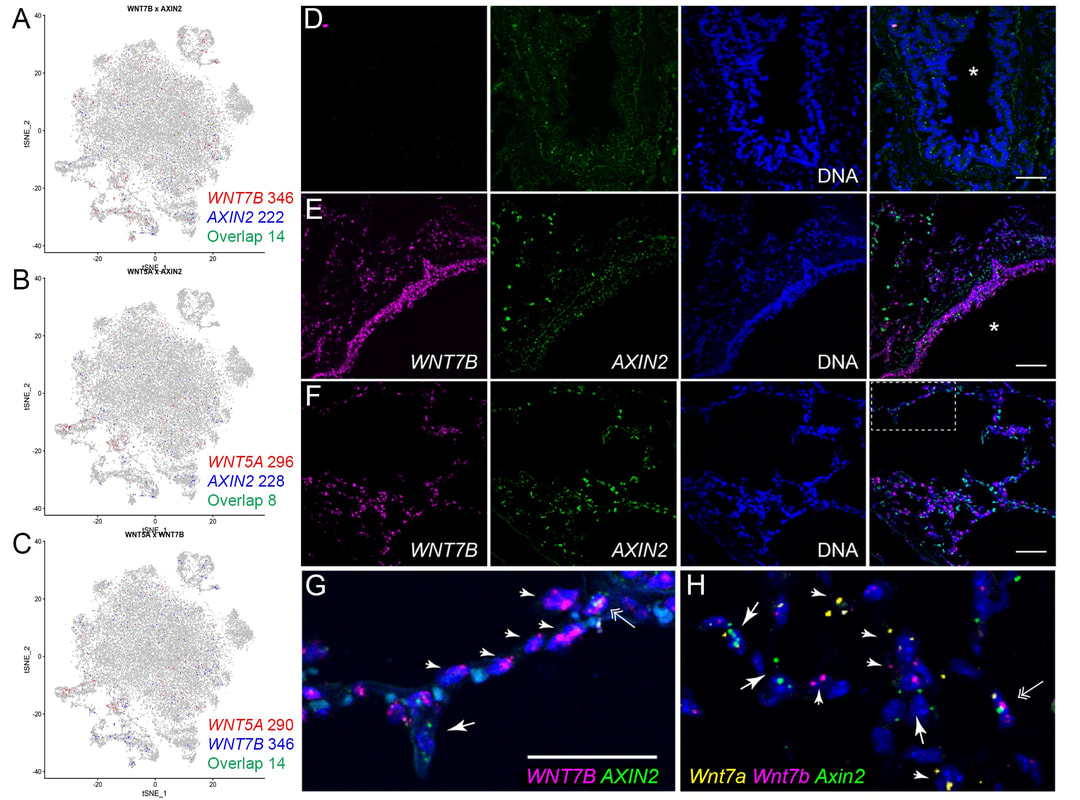
Nuclear roles for catenins: Wnt/beta-catenin signaling in fibrosis
When a tissue is injured, a coordinated multi-cellular response is required to repair the tissue. Fibroblasts play a critical role in the repair-response, but failure to keep these cells in check leads to persistent extracellular matrix secretion and fibrosis. Together with a team of collaborators from both Pulmonary and Rheumatology Divisions at Northwestern and Yale, we’ve found that too much nuclear signaling by b-catenin can drive tissue fibrosis, and that small molecular inhibitors that target the transcriptional activity of b-catenin may limit disease progression using mouse models for this disease. We are currently interested in the degree to which too much b-catenin signaling in monocyte-derived macrophages exacerbates the tissue repair response, and are actively trying to understand the tissue-resident signals that lead to sustained b-catenin signaling in these pathogenic macrophages, which may lead to the identification of additional therapeutic targets.
When a tissue is injured, a coordinated multi-cellular response is required to repair the tissue. Fibroblasts play a critical role in the repair-response, but failure to keep these cells in check leads to persistent extracellular matrix secretion and fibrosis. Together with a team of collaborators from both Pulmonary and Rheumatology Divisions at Northwestern and Yale, we’ve found that too much nuclear signaling by b-catenin can drive tissue fibrosis, and that small molecular inhibitors that target the transcriptional activity of b-catenin may limit disease progression using mouse models for this disease. We are currently interested in the degree to which too much b-catenin signaling in monocyte-derived macrophages exacerbates the tissue repair response, and are actively trying to understand the tissue-resident signals that lead to sustained b-catenin signaling in these pathogenic macrophages, which may lead to the identification of additional therapeutic targets.